
奇怪形状的峰代表了LC/HPLC实验室中可能出现的最常见问题之一 。首先应该解释为什么拖尾不好:
带有拖尾的峰很难整合
与良好的色谱法相比,涉及失真(带拖尾的峰)的分析方法的精确度和可靠性通常较差
当峰出现拖尾时分辨率低
峰拖尾的另一个原因是使用错误的溶剂溶解样品。
样品溶剂错误
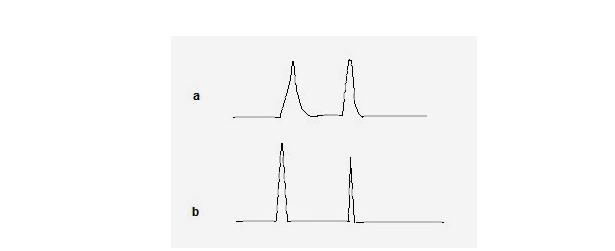
当用于溶解样品的体积和溶剂种类不合适时,会观察到峰拖尾。理想情况下,应注入少量样品(溶解在流动相中)。但是,当相对较大体积的比流动相强的溶剂与溶解的样品一起进样时,色谱图会受到严重影响。这显示在图1a,其中30 μ溶于乙腈的双组分的样品中的L 18%乙腈/水流动相被注入。出现两个拖尾和扭曲的峰。当相同的样品溶解在 18% 的乙腈/水中时,会出现两个正常峰。
将 30 毫升样品注入乙腈中。使用 18% 乙腈/水作为流动相的反相分离 b) 与 (a) 相同,但样品溶解在流动相中并出现 2 个正常峰。
从上面的讨论看来,选择溶解样品的溶剂应该使用以下程序:
最好的选择是将样品溶解在流动相中并注入 10-50 μ L
或者,在反相 LC 的情况下,可以注入更大体积的弱溶剂,即 100-500 μ L 溶解在水中的样品。一个主要的缺点是在色谱图的开头出现了更大的基线颠簸。
较大的体积(100-500 μ溶解在流动相中的样品L)也可使用,但分辨率可以特别苦为早期洗脱峰
在情况下,上述不工作,作为最后的手段10-25 μ比流动相更强的溶剂的L可被使用。
柱外效应
对于设计良好的 LC 系统,由于柱外体积导致条带拖尾的主要原因通常是检测器流通池。一些检测器允许替换较小体积的流通池,这会减少条带拖尾,但也会降低检测器的灵敏度。在某些情况下,连接管可能太长或其内径太大。可以用 0.007 英寸内径的管子代替它来解决这个问题。
LC 中的带前沿(图 2)可能是由柱温问题引起的,尤其是在环境温度下的离子对色谱中。在这种情况下,在较高温度 (~45 C) 下重复分离通常可以消除问题。40-50 C 的温度通常有利于离子对色谱,因为更窄的谱带和更好的分离结果。
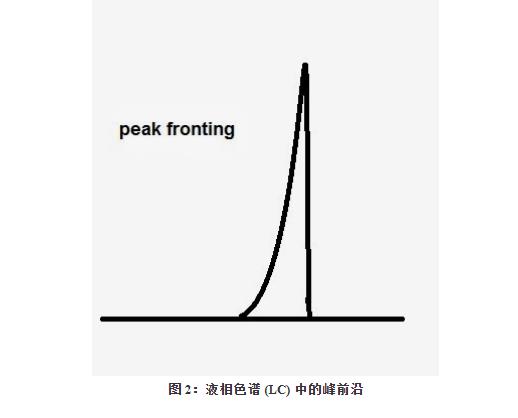
离子对色谱中前沿谱带的另一个来源是使用非流动相的样品溶剂。通常,在离子对色谱的样品-25〜30 μ升 -应该只被注入作为移动相的溶液,以避免带绕前和其他问题。
通过正相或离子交换色谱进行的分离涉及样品分子与柱填料表面的特定位点的结合。例如,阳离子交换剂上的磺酸盐 (-SO3 - ) 基团或二氧化硅上的硅烷醇 (Si-OH) 基团。通常,这些站点并不完全相同。一些位点位置优越,可与样品分子发生强相互作用。样品分子将优先选择最强的保留位点,并且将首先使用这些位点。因为这些强位点通常以低浓度存在,所以它们很快被样品占据,因此只有较弱的位点可用。
强保留位点的另一个特点是它们通常会吸引强保留的样品分子。这意味着过早的色谱柱过载主要发生在色谱图中较晚的洗脱条带上。因此,结果是色谱图中的最后一个条带显示条带拖尾。
这种由于强保留位点导致的离子交换或正相 LC 中的条带拖尾有时可以通过使用较少的样品来减少。更有效的方法是增加流动相的强度。
